
Rola oksygenazy hemowej w procesach zapalnych
Kamil Kruczek | 2009-08-19
Zapalenie (łac. inflammatio) jest szybkim, ale skoordynowanym procesem wywołanym przez zakażenie chorobotwórczymi drobnoustrojami lub uszkodzeniem tkanki. Główną funkcją zapalenia jest opanowanie i zakończenie infekcji lub naprawa zniszczonej tkanki i powrót do stanu homeostazy.
Istotna dla efektywności takiego systemu jest zdolność do wytworzenia szybkiej odpowiedzi adekwatnej do charakteru wyzwalającego czynnika z jednoczesnym maksymalnym ograniczeniem jej szkodliwych dla organizmu skutków. Stąd, idealna odpowiedź powinna charakteryzować się zarówno gwałtownością i destruktywnością (kiedy potrzeba), jak i specyficznością i samoograniczeniem. Waga tego zrównoważenia uwidacznia się w przypadku chronicznych infekcji lub schorzeń o podłożu zapalenia przewlekłego (1). Reakcje zapalne wyzwalane są przez aktywację komórek endotelialnych (EC – endothelial cells) oraz rezydujących w uszkodzonych tkankach makrofagów (Mø). Aktywacja EC powoduje migrację krążących leukocytów oraz związków rozpuszczalnych z krwi do tkanek. Zjawisko to opiera się na aktywacji ekspresji genów wczesnej odpowiedzi kodujących związki indukujące skurcz naczyń (endotelina-1), cytokiny (interleukiny: IL-1α, IL-6), chemokiny (IL-8) oraz cząsteczki adhezyjne (E i P-selektyny, intracellular adhesion molecule 1 (ICAM-1), vascular cellular adhesion molecule 1 (VCAM-1)), ich produkty mają własności prozapalne i stymulują migrację leukocytów. Aktywacja makrofagów natomiast powoduje uwalnianie prozapalnych cytokin (IL-1α/β, IL-6, TNF-α) oraz wzrost ekspresji genów prozapalnych np. cyklooksygenazy (COX-2), oksydazy NAD(P)H i indukowalnej syntazy tlenku azotu (iNOS), produkty tych genów powodują wytwarzanie wysokich stężeń odpowiednio prostaglandyn, wolnych rodników oraz tlenku azotu łącznie tworzących znaczny stres oksydacyjny wymierzony w komórki patogenu. Procesy te zachodzą po rozpoznaniu specyficznych wzorów molekularnych eksponowanych na powierzchni patogennych komórek (PAMPs – pathogen associated molecular patterns) i/lub cząsteczek wydzielanych przez zniszczone komórki (PRRs – pattern recognition receptors) (2,3). Zapalenie wiąże się zwykle także z aktywacją limfocytów T i B odpowiedzialnych za odporność nabytą opartą na rozpoznaniu specyficznych dla atakującego patogenu antygenów. Rozpoznanie antygenów przez komórki T może zajść tylko w kontekście głównego kompleksu zgodności tkankowej po ‘obróbce’ i ekspozycji na powierzchni komórek prezentujących antygen np. komórek dendrytycznych. Po rozpoznaniu tak zaprezentowanego antygenu następuje aktywacja komórek T której towarzyszą znaczne modyfikacje fenotypowe oraz logarytmiczne namnażanie w celu wytworzenia puli specyficznych antygenowo efektorowych limfocytów T mogących po migracji do miejsca infekcji eliminować atakujące drobnoustroje. W rozwoju procesu zapalnego wyróżnić można trzy etapy: początkowy, efektorowy oraz zakończenia. W fazie początkowej następuje wyżej wspomniana aktywacja EC, ekspresja chemokin, cytokin oraz cząsteczek adhezyjnych pośredniczących w rekrutacji krążących leukocytów, w fazie efektorowej następuje sekrecja cytokin prozapalnych, wolnych rodników, enzymów i związków skierowanych przeciwko drobnoustrojom. Dla uniknięcia nieodwracalnego uszkodzenia tkanek niezbędna jest faza zakończenia odpowiedzi zapalnej (4). Brak jej wystąpienia prowadzi do rozwoju chorób zapalnych, u podstaw których leżeć może zbyt niska aktywność genów przeciwzapalnych np. dyskutowanej oksygenazy hemowej.
Własności oksygenazy hemowej (HO) istotne dla stanów zapalnych
Oksygenaza hemowa (EC 1.14.99.3) jest pierwszym i ograniczającym enzymem na szlaku degradacji hemu, generującym równomolowe ilości biliwerdyny IXα , wolnego jonu żelaza (II) oraz tlenku węgla. Następnie biliwerdyna jest szybko przekształcana do bilirubiny przez reduktazę biliwerdyny, natomiast wolny jon Fe2+ jest wyłapywany przez ferrytynę (5). Produkty degradacji hemu są ważnymi związkami biologicznie aktywnymi. CO wiąże się z hemoglobiną z wytworzeniem karboksyhemoglobiny, w tej postaci jest transportowany do płuc, gdzie jest wydzielany w wydychanym powietrzu. CO zyskał wiele uwagi ze względu na fizjologiczną rolę zbliżoną do tlenku azotu (NO). Biologiczne znaczenie CO przejawia się w jego właściwościach przeciwzapalnych antyapoptotycznych oraz hamujących proliferację komórek (6). Rozpoznano trzy geny dla izoform HO: ho-1, ho-2 oraz słabo scharakteryzowany ho-3, najprawdopodobniej pseudogen nie kodujący funkcjonalnego białka (4). Z dwóch funkcjonalnych izoform (HO-1 i HO-2) HO-1 jest wysoce indukowalna i ulega ekspresji w wielu typach tkanek w wyniku działania różnorodnych bodźców stymulujących takich jak hem i inne oksydanty, endotoksyna, zapalenie, cytokiny prozapalne, hipoksja, hiperoksja oraz metale ciężkie. HO-2 natomiast jest syntetyzowane konstytutywnie i ulega ekspresji w wielu organach, szczególnie mózgu i gonadach (5). Indukcja HO-1 najprawdopodobniej przedstawia mechanizm obronny przeciw stresowi oksydacyjnemu. Relatywnie stały poziom ekspresji HO-2 może być odpowiedniejszy dla funkcji regulacyjnej HO-2 w homeostazie tlenowej. HO-2 prawdopodobnie pełni funkcję sensoryczną dla stężenia tlenu na poziomie komórek mięśni gładkich tętnicy płucnej odpowiedzialnych za dopasowanie wentylacji-perfuzji do zoptymalizowania utlenowania krwi w krążeniu płucnym (6).
Mechanizmy działania HO w stanach zapalnych
Hem jest cząsteczką potencjalnie niebezpieczną po uwolnieniu z wewnątrzkomórkowych hemoprotein. Cytotoksyczne efekty hemu objawiają się przez wytwarzanie stresu oksydacyjnego poprzez generowanie reaktywnych form tlenu (ROS). Mechanizm generacji ROS opiera się na reakcji Fentona. W wyniku tych procesów dochodzi do peroksydacji lipidów i rozwoju reakcji zapalnej (7). Hem katalizuje również peroksydację podatnych grup białek błonowych, białek osocza oraz białek cytozolowych, prowadząc do formowania kowalencyjnych wiązań krzyżowych bądź fragmentacji polipeptydów (8). Degradacja wolnego hemu prowadzi więc do obniżenia stresu oksydacyjnego i obniżenia odpowiedzi zapalnej. W czasie reakcji zapalnych poziom ekspresji HO-2 nie ulega większym zmianom, silnej indukcji ulega natomiast HO-1. Przypuszcza się, iż w tych warunkach HO-1 działa jako enzym ochronny obniżający ekspresję prozapalnych produktów genów związanych z aktywacją EC i Mø oraz chroniący nielimfoidalne komórki przed apoptozą (4). Udział HO-1 w procesach zapalnych potwierdza analiza fenotypu jedynego pacjenta z wrodzonym brakiem HO-1 charakteryzującego się m. in. nasiloną wrażliwością na uszkodzenia związane ze stresem oksydacyjnym oraz syndromem zapalnym, będącym przyczyną śmierci w wieku 6 lat (7).
Rozwinięty stan zapalny charakteryzujący się znacznym stresem oksydacyjnym powoduje reakcję wolnych rodników z powstałą z hemolizy erytrocytów hemoglobiną z wytworzeniem prozapalnej methemoglobiny. Methemoglobina po związaniu z białkiem ostrej fazy haptoglobiną jest rozpoznawana przez receptory HbSr/CD163 na powierzchni Mø (9), co wywołuje indukcję HO-1 oraz wydzielanie CO, który blokuje prozapalny TNF-α i stymuluje sekrecję przeciwzapalnej IL-10. Efekt przeciwzapalny IL-10 jest w mysich makrofagach zależny od indukcji HO-1 i zachodzi przez szlak zależny od aktywowanej mitogenem kinazy białkowej p38 (MAPK) (10). Wzrost aktywności HO-1 jest prawdopodobny w Mø internalizujących hemoglobinę poprzez HbSr/CD163, co zapewnia substrat dla enzymu niezbędny do wytwarzania CO. Sugeruje to, iż makrofagi eksponujące receptor HbSr/CD163 wykazują głównie aktywność przeciwzapalną promującą zakończenie reakcji zapalnej (Schemat 1). Również aktywność innych cząsteczek przeciwzapalnych opiera się na modulacji aktywności HO-1, dzieje się tak także w przypadku 15-deoksy-Δ12,14-prostaglandyny J2 (15d-PGJ2), cyklopentanonowej prostaglandyny wykazującej silne działanie przeciwzapalne oparte prawdopodobnie na indukcji wytwarzania CO przez HO-1 (4). Traktowanie Mø aktywowanych lipopolisacharydem (LPS) i interferonem gamma (IFN-γ) 15d-PGJ2 powoduje inhibicję zależnych od tych czynników genów, zahamowanie wytwarzania NO. Działanie 15d-PGJ2 polega na inhibicji degradacji inhibitora NF-κB (IκB), co powoduje obniżenie aktywności NF-κB oraz ekspresji zależnych od tego czynnika genów m. in. COX-2 i iNOS (11). Podobnie flawonoidy takie jak aglikony hesperetyna (HT) i narigenina (NE) działają poprzez indukcję HO-1 i inhibicję aktywności iNOS (a tym samym wytwarzania NO) (12). Obserwacje te pozwalają na przypuszczanie, iż HO-1 może pełnić w regulacji procesów zapalnych funkcję ‘zlewu’ integrującego działanie wielu czynników przeciwzapalnych (4).
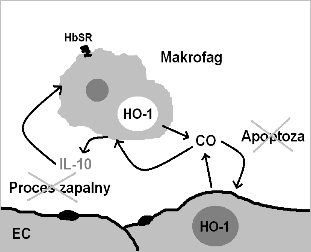
Schemat 1. Komórkowe efekty działania CO: ochrona EC przed apoptozą, modulacja prozapalnego fenotypu aktywowanych makrofagach, pętla sprzężenia zwrotnego pomiędzy IL-10 i HO-1 (na podstawie (4)).
W komórkach EC nie ulega ekspresji receptor HbSr/CD163, stąd ich większa wrażliwość na obecność w otoczeniu prozapalnej (met)hemoglobiny. Hem/(met)hemoglobina indukuje w EC ekspresję cząsteczek adhezyjnych (E i P-selektyn, VCAM-1, ICAM-1) (8). Poprzez CO i szlak p38 MAPK HO-1 chroni EC przez apoptozą wywołaną rozwojem reakcji zapalnej (13). Rola HO-1 w aktywacji EC widoczna jest po podaniu specyficznego inhibitora - cynkowej protoporfiryny IX (ZnPP IX), w wyniku czego dochodzi do wzmocnienia migracji, toczenia i adhezji neutrofili na EC. Przeciwne efekty wywierają substrat HO hemina, donory CO oraz biliwerdyna (BVD). Inhibicja migracji neutrofili przez NO jest również zależna od aktywności HO-1 (14). HO-1 jest także istotnym składnikiem pętli ujemnego sprzężenia zwrotnego w aktywacji/proliferacji CD4+ limfocytów T, a jej działanie odbywa się poprzez inhibicję ekspresji czynników prozapalnych (np. TNF-α) oraz CO wykazujący działanie proapoptotyczne w stosunku do limfocytów T przez receptor Fas95/CD95 (4). Ten efekt proapoptotyczny przeciwny do wywieranego na EC może być pomocy w zakończeniu odpowiedzi zapalnej i ograniczeniu uszkodzeń tkanek. Niedostateczna indukcja HO-1 w warunkach stresu oksydacyjnego powodującego uszkodzenia tkanek prowadzi do rozwoju schorzeń o podłożu zapalnym. Znajduje to potwierdzenie w badaniach rozwoju przewlekłej obturacyjnej choroby płuc (COPD) wywołanej ekspozycją na dym papierosowy. W patogenezie schorzenia istotną rolę odgrywają nacieki limfocytarne głównie kom. B oraz CD4+CD25+ limfocytów T oraz wysoki poziom prozapalnych cytokin uwalnianych przez komórki immunologiczne. Indukcja HO-1 kobaltową protoporfiryną IX powoduje znaczne zmiejszenie infiltracji dotkniętych tkanek przez kom. B i CD4+CD25+ T (15). U myszy transgenicznych ze wzmocnioną ekspresją HO-1 widoczne jest podobne zmniejszenie nacieków limfocytarnych w myocardium poddanym ischemii/reperfuzji (5). Obserwacje te pozwalają wnioskować o wpływie indukcji HO-1 na modulację aktywności i migracji również limfocytów B.
Mechanizmy działania produktów aktywności HO-1: Fe, CO i BVD
Przeciwzapalna aktywność HO-1 wyraża się przez obniżenie komórkowego stężenia wolnego Fe, np. w aktywowanych EC, prowadzi to inhibicji NF-κB oraz zależnych od niego genów a także hamując bezpośrednio biosyntezę hemoprotein biorących udział w regulacji procesów zapalnych np. oksydazy NAD(P)H, COX-2 oraz iNOS. Ponadto aktywność biologiczną wykazują CO i BVD. CO może się wiązać z Fe grup hemowych i klasterów Fe-S oraz dyfundować przez błonę poza komórkę, gdzie może związać się z hemoglobiną erytrocytów i zostać przetransportowany do płuc i wydzielony z wydychanym powietrzem (4). Wiązanie do hemoprotein może modulować ich aktywność biologiczną, co ma miejsce w przypadku cyklazy guanylowej, związanie CO ma zdolność, podobnie do NO, stymulacji produkcji cGMP przez enzym (4,8). Dodatkowo CO może wywierać poprzez szlak p38 MAPK swe efekty antyapoptotyczne i przeciwpodziałowe w EC, Mø i komórka mięśni gładkich (4,16).Niejasny pozostaje mechanizm efektów antyapoptotycznych na EC. Zaobserwowano, iż aktywacja p38 MAPK prowadzi do inhibicji efektorowych kaspaz-8 i -3 a CO może hamować ekspresję Fas/Fas-ligandu, blokować aktywację kaspaz-3, -8 i –9 a także uwalnianie cytochromu c z mitochondriów, które to procesy są kluczowe dla inicjacji apoptozy w tych komórkach. W komórkach mięśni gładkich aktywacja p38 MAPK przez CO zachodzi przez cyklazę guanylową i akumulację cGMP, wzrasta także ekspresja p21cip1/waf, inhibitora cyklu komórkowego modulującego aktywność NF-κB. W tych komórkach CO również wywiera efekty antyapoptotyczne (4). Badania na mutantach myszy z knockoutami różnych kinaz MAP sugerują że specyficznej aktywacji przez CO ulega szlak MKK3/p38β MAPK (17). Niedawne badania wskazują na nowy mechanizm wpływu CO na komórki endotelialne poprzez aktywację wysokoprzepływowych kanałów potasowych (BK). Aktywacją BK odbywa się zarówno mechanizmem bezpośrednim, jak i poprzez szlak zależny od akumulacji cGMP i wytwarzania NO (18), prowadzi to do relaksacji naczyń.
Biliwerdyna i bilirubina były uważane jedynie za odpadowe produkty metabolizmu hemu, toksyczne w wysokich stężeniach, szczególnie u noworodków, do czasu obserwacji, iż bilirubina wykazuje wyższe od α-tokoferolu własności antyoksydacyjne w ochronie lipidów błonowych przed peroksydacją. Własności te mogą zapewniać zabezpieczenie prze oksydacją LDL i spowalniać w ten sposób rozwój płytek miażdżycowych (8). Utlenione metabolity bilirubiny są wydalane w moczu po ekspozycji na stres oksydacyjny (8).
Przegląd znaczenia w HO-1 w chorobach zapalnych
Układ krwionośny. Myszy transgeniczne z nadekspresją HO-1 wykazują poprawę funkcji serca, zmniejszony obszar martwicy mięśnia sercowego w zawale, obniżone uszkodzenia zapalne i oksydacyjne po ligacji tętnicy wieńcowej i reperfuzji. Stan zapalny leży u podłoża rozwoju miażdżycy. Depozycja lipidów w ścianach naczyń związana z podniesionym poziomem cholesterolu we krwi prowadzi do tworzenia płytek miażdżycowych, proces wiąże się w pobieraniem utlenionej formy lipoproteiny niskiej gęstości (oxidized low density lipoprotein; oxLDL). Wysoki poziom aktywności HO-1 hamuje chemotaksję monocytów in vitro oraz peroksydację lipidów i obniża stres oksydacyjny przez co przyczynia się do redukcji tworzenia płytek miażdżycowych (5,8).
Układ oddechowy. W płucach HO-1 ulega ekspresji w różnych typach komórek m. in. pneumocytach typu II i makrofagach pęcherzykowych a aktywność enzymu indukowana jest przez hem, hipoksję, hiperoksję, NO, endotoksynę oraz cytokiny prozapalne. Poziom ekspresji HO-1 jest podwyższony w wielu chorobach układu oddechowego o podłożu zapalnym m. in. zespole ostrej niewydolności oddechowej (acute respiratory distress syndrome; ARDS), COPD, astmie, mukowiscydozie (cystic fibrosis; CF) oraz reakcjach odrzucenia po transplantacji płuca (19,6) W mysim modelu astmy ekspresja HO-1 jest podwyższona w świetle dróg oddechowych i warstwie podśluzówkowej oskrzeli w poddanych działaniu antygenu zwierzętach. Aktywacja HO-1 skutkuje obniżeniem nasilenia reakcji zapalnych, wydzielania śluzu i reaktywności na histaminę. W próbkach plwocin pacjentów z astmą wykrywa się podwyższoną zawartość bilirubiny oraz stężenie CO w wydychanym powietrzu, co świadczy o podobnej aktywacji HO-1 jak w modelu zwierzęcym (6) W COPD natomiast nadekspresja HO-1 wywołana transgenem w wektorze adenowirusowym obniżała nasilenie zapalenia, stężenie cytokin prozapalnych i uszkodzenia tkanek wywołane przez wolne rodniki generowane przez dym tytoniowy poprzez inaktywację inhibitorów proteaz, której towarzyszy niszczenie macierzy pozakomórkowej (19). W progresji CF także znaczący udział ma generacja ROS i indukcja HO-1, której poziom może być miernikiem nasilania objawów chorobowych, a nadekspresja HO-1 zmniejsza uszkodzenia komórek epitelium dróg oddechowych (19).
Układ nerwowy. Stwardnienie rozsiane (multiple sclerosis; MS) uważa się za chorobę autoimmunologiczną, progresja schorzenia charakteryzuje się występowaniem płytek neurozapalnych powstających w wyniku interakcji aktywowanych patogennych kom. Th z komórkami prezentującymi antygen (APCs) w centralnym układzie nerwowym (CNS). Prowadzi to do odpowiedzi zapalnej skutkującej nieodwracalnymi uszkodzeniami oligodendrocytów, demielinacją neuronów i, ostatecznie, utratą aksonów. W mysim modelu MS – eksperymentalnym autoimmunologicznym zapaleniu mózgu (experimental autoimmune encephalomyelitis (EAE)) – ekspresja HO-1 zapobiega rozwojowi zapalenia, demielinacji i paraliżowi oraz stymuluje remisję objawów chorobowych (20). Podobne znaczenie HO-1 może wykazywać w patologii innych chorób neurodegeneracyjnych związanych z reakcjami zapalnymi m. in. chorobie Alzheimera oraz chorobie Parkinsona w przypadku której podwyższona ekspresja HO-1 została zaobserwowana w substancji czarnej pacjentów z idiopatycznym wariantem choroby (6).
Choroby zakaźne. HO-1 pełni istotną funkcję w odpowiedzi przeciwko patogenom w rozwoju sepsy. Myszy z delecją HO-1 wykazują podwyższoną śmiertelność w wyniku wielobakteryjnej sepsy. Podwyższona ekspresja HO-1 nie powoduje supresji krążących kom. immunologicznych ani nie hamuje ich akumulacji w miejscu zakażenia, ale wzmaga usuwanie patogenów m. in. przez ich wzmożoną fagocytozę (21). Również w przebiegu malarii modulacji ulega poziom ekspresji HO-1. Podniesione stężenie wolnego hemu w organach dotkniętych chorobą sprzyja zapaleniu naczyń krwionośnych i stymuluje HO-1. Traktowanie chorych myszy heminą nasila ekspresję HO-1, co hamuje związane z występującymi w chorobie niedrożnościami naczyń uszkodzenia waskularnego endotelium w wyniku hipoksji/reperfuzji, interakcje leukocytów z endotelium oraz ekspresję NF-κB, VCAM-1 i ICAM-1.
Podsumowanie
Poziom ekspresji HO-1 ma istotne znaczenie w przebiegu reakcji zapalnych leżących u podłoża wielu chorób. Wiele z mechanizmów działania HO-1 i modulacyjnej aktywności produktów tego enzymu pozostaje do wyjaśnienia. Rozwianie pozostałych niepewności pozwoli na dokładniejsze poznanie regulacji procesów zapalnych, jak i będzie niosło potencjalne możliwości zastosowania w terapii chorób zapalnych.
Kamil Kruczek
kamil.kruczek@uj.edu.pl
Literatura:
(1)Barton G. M. A calculated response: control of inflammation by the innate immune system. Clin Invest. 2008 February 1; 118(2): 413–420.
(2)Hoffman JA., Kafatos FC, Janeway CA, Ezekowitz RA. Phylogenetic perspectives in innate immunity. Science1999 May 21;284(5418):1313-8.
(3)Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature 2007 Oct 18;449(7164):819-26.
(4)Silva G., Gregoire I., Tokaji L., Chora A., Seldon M., Cavalcante M., Soares M. Heme Oxygenase-1: A protective Gene that Regulates Inflammation and Immunity Heme Oxygenase 2005 Nova Science
(5)Perrella M., Shaw-Fang Y. Heme Oxygenase-1 in Cardiovascular Biology Heme Oxygenase 2005 Nova Science
(6)Yamaya M., Shibahara S. Heme Oxygenase and Human Disease Heme Oxygenase 2005 Nova Science
(7)Hyun-Ock Pae, Eun-Cheol Kim, Hun-Taeg Chung Integrative Survival Response Evoked by Heme Oxygenase-1 and Heme Metabolites J Clin Biochem Nutr. 2008 May; 42(3): 197–203
(8)Morita T. Heme Oxygenase and Atherosclerosis Arterioscer. Thromb. Vasc. Biol. 2005;25;1786-1795.
(9)Fagoonee S., Gburek J., Hirsch E., Marro S., Moestrup S., Laurberg J., Christensen E., Silengo L., Altruda F., Tolosano E. Plasma Protein Haptoglobin Modulates Renal Iron Loading Am J Pathol. 2005 April; 166(4): 973–983.
(10)Lee TS., Chau LY. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice Nat Med. 2002 Mar;8(3):240-6.
(11)Castrillo A., Díaz-Guerra M., Hortelano S., Martín-Sanz P., Boscá L. Inhibition of IκB Kinase and IκB Phosphorylation by 15-Deoxy-Δ12,14-Prostaglandin J2 in Activated Murine Macrophages Mol Cell Biol. 2000 March; 20(5): 1692–1698.
(12)Lin HY, Shen SC, Chen YC. Anti-inflammatory effect of heme oxygenase 1: glycosylation and nitric oxide inhibition in macrophages. J Cell Physiol. 2005 Feb;202(2):579-90.
(13)Brouard S., Otterbein L., Anrather J., Tobiasch E., Bach F., Choi A., Soares M. Carbon Monoxide Generated by Heme Oxygenase 1 Suppresses Endothelial Cell Apoptosis J Exp Med. 2000 October 2; 192(7): 1015–1026.
(14)Freitas A., Alves-Filho A., Secco D., Neto A., Ferreira S., Barja-Fidalgo C., Cunha F. Heme oxygenase/carbon monoxide-biliverdin pathway down regulates neutrophil rolling, adhesion and migration in acute inflammation. J Clin Invest. 2007 February 1; 117(2): 438–447.
(15)Brandsma C., Hylkema M., van der Strate B., Slebos D., Luinge M., Geerlings M., Timens W., Postma D., Kerstjens H. Heme oxygenase-1 prevents smoke induced B-cell infiltrates: a role for regulatory T cells? Respiratory Research 2008, 9:17 doi:10.1186/1465-9921-9-17
(16)Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, Davis RJ, Flavell RA, Choi AM. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med. 2000 Apr;6(4):422-8
(17)Otterbein L., Otterbein S., Ifedigbo E., Liu F., Morse D., Fearns C., Ulevitch R., Knickelbein R., Flavell R., Choi A. MKK3 Mitogen-Activated Protein Kinase Pathway Mediates Carbon Monoxide-Induced Protection Against Oxidant-Induced Lung Injury. Am J Pathol. 2003 December; 163(6): 2555–2563.
(18)Dong DL, Zhang Y, Lin DH, Chen J, Patschan S, Goligorsky MS, Nasjletti A, Yang BF, Wang WH. Carbon monoxide stimulates the Ca2(+)-activated big conductance k channels in cultured human endothelial cells. Hypertension. 2007 Oct;50(4):643-51. Epub 2007 Aug 27.
(19)Fredenburgh L., Perrella M., Mitsialis A. TheRole of Heme Oxygenase-1 in Pulmonary Disease Am. J. Respir. Cell Mol. Biol. Vol. 36 pp 158-165, 2007
(20)Chora A., Fontoura P., Cunha A., Pais T., Cardoso S., Ho P., Lee L., Sobel R., Steinman R., Soares M. Heme oxygenase–1 and carbon monoxide suppress autoimmune neuroinflammation. J Clin Invest. 2007 February 1; 117(2): 438–447.
(21)Chung S., Liu X., Macias A., Baron R., Perrella M. Heme oxygenase-1–derived carbon monoxide enhances the host defense response to microbial sepsis in mice. J Clin Invest. 2006 March 1; 116(3): 808–816.